
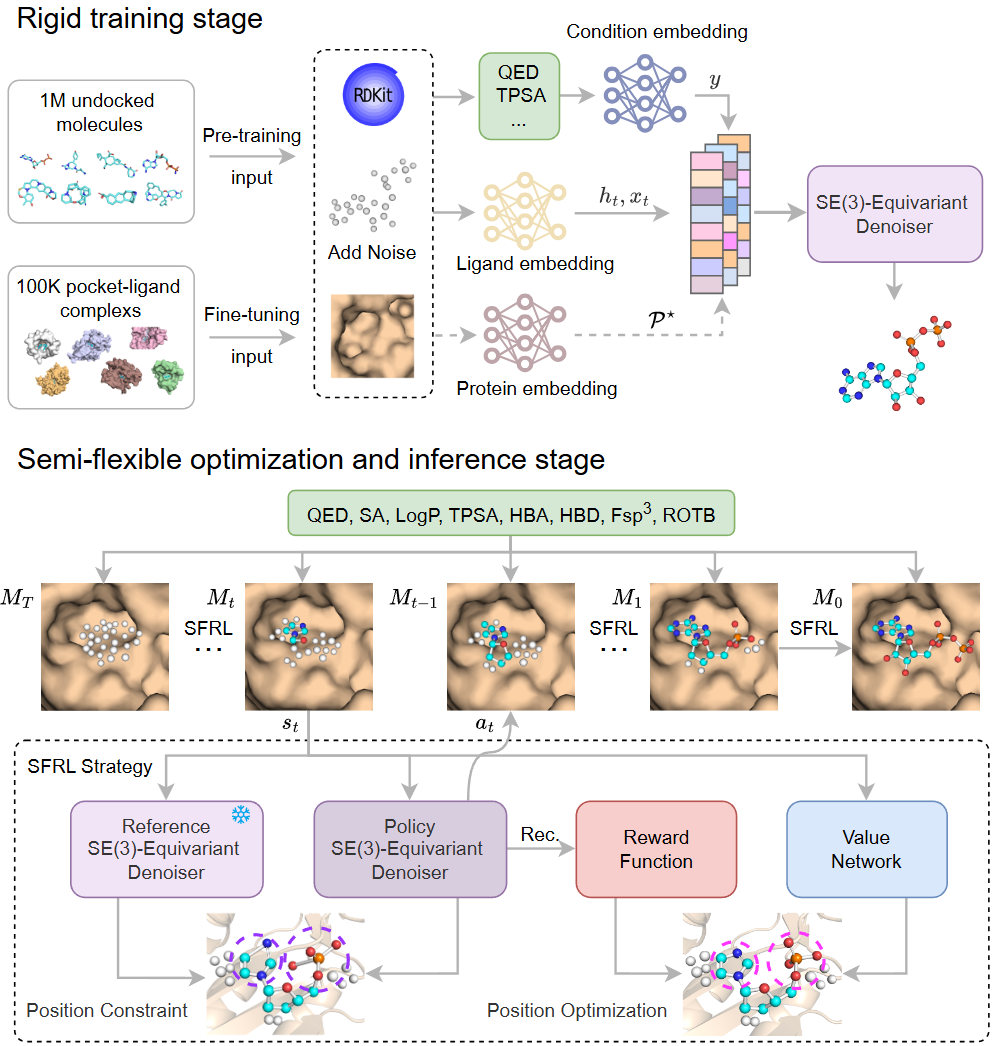
SeFMol is designed for structure-based drug design, where the goal is to generate 3D ligand molecules directly inside a protein binding pocket. Unlike methods that treat ligands as rigid during training, SeFMol is inspired by semi-flexible docking and allows molecular conformations to be adjusted during the denoising process, making the generated candidates more compatible with pocket geometry and interaction patterns.
What makes SeFMol different
- Generates 3D molecules conditioned on a protein pocket.
- Uses reinforcement learning to steer semi-flexible conformational optimization.
- Supports molecular property guidance such as QED, SA, LogP, TPSA, HBA, HBD, Fsp3, and ROTB.
What users can expect
- Candidate molecules with strong docking-related performance.
- Better control over drug-like physicochemical properties.
- Fast sampling suitable for practical lead exploration and early candidate triage.
Two-stage rigid training
- Pretraining: 1,000,000 target-free molecules from Molecule3D.
- Fine-tuning: 100,000 protein-ligand pairs from CrossDocked2020.
- Property guidance uses 8 RDKit-calculated properties: QED, SA, LogP, TPSA, HBA, HBD, Fsp3, and ROTB.
SFRL: semi-flexible RL optimization
- The denoising process is formulated as a Markov Decision Process (MDP).
- A policy denoiser optimizes molecular states step by step inside the target pocket.
- KL regularization constrains policy drift from the pretrained denoiser.
- PPO-style clipping and a value function are used to stabilize optimization.
Upload Target Structure
Provide the target protein structure and make sure the binding pocket is meaningful and complete.
Set Property Guidance
Choose the sample number and optionally specify target molecular properties for generation.
Run Generation
Launch SeFMol inference to generate 3D molecules under pocket and property constraints.
Inspect and Prioritize
Review structures, docking-related scores, and property values to identify promising candidates.
Required input
- Protein structure / pocket information used as the spatial condition for generation.
- Use a clean and chemically meaningful structure whenever possible.
- Binding-site geometry should be relevant to the design objective.
Optional guidance
- Property targets for QED, SA, LogP, TPSA, HBA, HBD, Fsp3, and ROTB.
- Sample count to control how many candidate molecules are generated.
- If you are unsure where to start, use the default property vector from the paper.
| Parameter | Meaning | Reference Value / Range | Interpretation Guidance |
|---|---|---|---|
| QED | Quantitative estimate of drug-likeness | default 1.0; SR criterion > 0.25 | Higher values usually indicate a more drug-like overall profile. |
| SA | Synthetic accessibility | default 1.0; SR criterion > 0.59 | Useful for checking whether generated molecules remain practically synthesizable. |
| LogP | Hydrophobicity balance | default 1.0; commonly -0.4 to 5.6 | High values may help permeability but can reduce solubility. |
| TPSA | Topological polar surface area | default 50.0; often < 90, SR criterion ≤ 140 | Important for polarity, membrane transport, and exposure behavior. |
| HBA | Hydrogen-bond acceptors | default 3.0; usually ≤ 10 | Helps tune intermolecular interaction patterns and polarity. |
| HBD | Hydrogen-bond donors | default 2.0; usually ≤ 5 | Useful when balancing binding interactions and developability. |
| FSP3 | 3D saturation level | default 0.5; typically > 0.47, SR criterion ≥ 0.42 | Higher values often improve 3D character and scaffold richness. |
| ROTB | Rotatable bonds | default 2.0; usually ≤ 10 | Lower values often help conformational stability. |
| num_samples | Number of generated molecules | paper evaluation: 100 per pocket | More samples improve coverage, but also increase screening workload. |
| Additional reported indicators | Value | Notes |
|---|---|---|
| Fast sampling | 1000 → 50 steps | About 20× acceleration during sampling. |
| Test scale | 100 protein pockets | Used in the benchmark evaluation. |
| Sampling per pocket | 100 molecules | Used for model comparison. |
| Interaction-pattern JSD | 0.1401 | Best reported value, tied with TargetDiff. |
| Case studies | CDK2 / ROCK1 | SeFMol reproduced known interactions and explored new ones. |
| Generalization | AlphaFold structures | Also showed favorable Vina score distributions on predicted proteins. |
These are paper-reported results intended to describe the method’s performance. Actual web runs may vary across targets, pocket quality, and parameter settings.
- Use biologically meaningful and structurally clean binding pockets.
- Start with the default property vector, then adjust one or two properties at a time.
- Do not screen candidates by Vina score alone; combine affinity, QED, SA, TPSA, and Fsp3.
- Cluster generated molecules before detailed review to reduce redundant chemotypes.
- Pair SeFMol with downstream docking, ADMET, and synthesis-feasibility tools for better triage.
- Keep records of inputs, parameters, and output files for reproducibility.
Xudong Zhang, Sanqing Qu, Fan Lu, Jianmin Wang, Zhixin Tian, Shangding Gu, Yanping Zhang, Alois Knoll, Shaorong Gao, Guang Chen, Changjun Jiang, Steering Semi-Flexible Molecular Diffusion Model for Structure-Based Drug Design with Reinforcement Learning.
Code and resources: https://github.com/ispc-lab/SeFMol